Bệnh Moyamoya
- Giới thiệu:
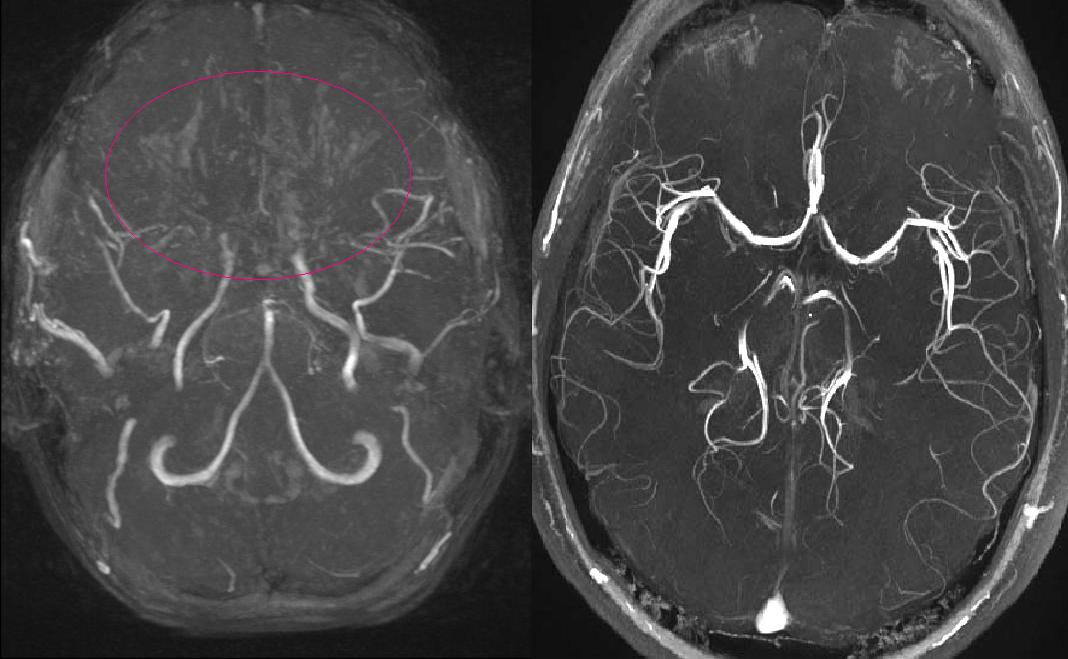
Bệnh Moyamoya là một bệnh mạch máu não độc đáo được đặc trưng bởi sự thay đổi hẹp tiến triển mãn tính nguyên phát của phần cuối của động mạch cảnh trong hai bên. Những thay đổi này gây ra sự hình thành một mạng lưới mạch máu bất thường là các con đường phụ để bù đắp cho thiếu máu cục bộ não liên quan đến sự thay đổi bệnh lý nguyên phát. Mạng lưới mạch máu hình thành thứ phát này được gọi là các “mạch máu moyamoya”.
Thuật ngữ “moyamoya” trong tiếng nhật có nghĩa là đám khói, được sử dụng để mô tả sự xuất hiện của các mạch phụ này trên chụp mạch.
Bệnh moyamoya được mô tả ca bệnh đầu tiên vào năm 1957 bởi Takeuchi và Shimizu, sau đó được Jiro Suzuki đặt tên năm 1969.
Biểu hiện lâm sàng của moyamoya trên hầu hết các bệnh nhân nhi khoa đều có các đợt thiếu máu cục bộ từ thiếu máu cục bộ thoáng qua (TIA) cho đến các cơn nhồi máu tiến triển. Ở các bệnh nhân nhi, TIA và nhồi máu có thể được gây ra bởi các hoạt động như thổi thức ăn nóng hay thổi nhạc cụ…
Ở người lớn, một nửa số trường hợp bị xuất huyết nội sọ.
- Dịch tễ
Tỉ lệ mắc bệnh moyamoya có liên quan đến yếu tố chủng tộc. Mặc dù phân bố của nó là trên toàn thế giới, tuy nhiên, chủ yếu xuất hiện ở người Đông Á.Một cuộc khảo sát dịch tễ học của Nhật bản được thực hiện bởi Wakai và các cộng sự (1994) tỷ lệ lưu hành bệnh là 3,16 trên 100.000 dân với tỷ lệ mắc mới 0,35 trên 100.000 dân.
Các nghiên cứu từ Hoa Kỳ cho thấy tỷ lệ mắc là 0,086 trên 100.000 dân. So với người da trắng, tỉ lệ mắc của người Mỹ gốc Á gấp 4.6 lần, người Mỹ gốc Phi gấp 2,2 lần và người Mỹ gốc Tây Ban Nha gấp 0,5 lần.
Theo một cuộc khảo sát gần đây ở Nhật Bản (khu vực Hokkaido), số lượng bệnh nhân mắc bệnh moyamoya tăng rõ rệt so với một cuộc khảo sát quốc gia trước đó. Sự gia tăng này có thể phản ánh sự gia tăng phát hiện về căn bệnh này và sự xuất hiện của nó mang tính gia đình.
Tỷ lệ mắc bệnh giữa nam/nữ là ½
- Sinh bệnh học
Sinh bệnh học của bệnh moyamoya chưa rõ ràng. Tỉ lệ gặp cao ở Nhật Bản và châu Á, cùng với tỉ lệ bệnh nhân có yếu tố gia đình lên đến khoảng 10%, một gợi ý lớn tới sinh bệnh học mang tính chất gen.
Các bằng chứng cho đến nay cho thấy rằng gen RNF213 trên nhiễm sắc thể 17425.3 là một tác nhân nhạy cảm quan trọng với MMD ở cộng đồng châu Á. Các nghiên cứu khác chỉ ra MMD có tính chất gia đình liên quan đến các nhiễm sắc thể 3p24.2-p26; 6q25; 8q23 và 12q12.
- Triệu chứng lâm sàng
Biểu hiện của bệnh lý moyamoya rất đa dạng và phụ thuộc vào 2 cơ chế chính

- Hội chứng Moyamoya (moyamoya thứ phát): bệnh moyamoya kèm với các bênh lý thường gặp dưới dây:
- Xơ vữa động mạch
- Nhiễm trùng: viêm màng não; viêm nhiễm do vi khuẩn hoặc virus
- Bệnh lý máu: hồng cầu hình liềm, beta thalassemia, lupus ban đỏ
- Tổn thương mô liên kết và hội chứng thần kinh da: NF1, củ lao, Hc Sturge – weber
- Bệnh chuyển hoá: tăng phosphate, nhiễm độc oxalat
- Bệnh lý thận đa nang
- Cận lâm sàng
Chụp CT và/hoặc MRI não là các xét nghiệm quan trọng trong việc phát hiện ra nhồi máu và chảy máu não ở bệnh moyamoya. Các phương pháp chụp mạch não không can thiệp và có can thiệp có thể xác định được tình trạng hẹp hoặc tắc của đa giác Willis. Siêu âm xuyên sọ là một phương pháp không xâm lấn để đánh giá huyết động nội sọ và tình trạng hẹp các động mạch lớn. Một số các phương pháp có thể được sử dụng để đánh giá tưới máu não khi nghỉ và sự đảo ngược dòng máu.

Hình 1: Hình ảnh ổ nhồi máu não vị trí thuỳ trán phải
 Hình 2: xuất huyết bên trong não và não thất.
Hình 2: xuất huyết bên trong não và não thất.

Hình 3: hình ảnh chụp mạch của một bệnh nhi 3 tuổi cho thấy hình ảnh hẹp đoạn cuối động mạch cảnh trong trái và đoạn gần động mạch não trong phải. Bên cạnh đó có sự hình thành các đám mạch máu Moyamoya.
- Chẩn đoán
Lưu đồ chẩn đoán bệnh lý Moya-moya

Chẩn đoán giai đoạn bệnh
|
GĐ |
Đặc điểm trên chụp mạch |
|
1 |
Hẹp chỗ chia đôi động mạch cảnh trong đơn thuần |
|
2 |
Khởi đầu của MMD ở nền với sự dãn ra của tất cả các động mạch não chính |
|
3 |
MMD diễn biến nặng hơn với sự giảm lưu lượng của động mạch não trước và não giữa |
|
4 |
Các mạch máu nhỏ bị moyamoya; phần trung tâm của động mạch não sau bị tổn thương |
|
5 |
Giảm moyamoya và không còn hình ảnh của các động mạch não chính |
|
6 |
Các mạch moyamoya bị biến mất; tuần hoàn não chỉ được cấp máu bởi hệ động mạch cảnh ngoài |

Hình 4: Các giai đoạn của bệnh lý Moyamoya
- Điều trị
7.1. Nội khoa:
Không thể phục hồi/ đảo ngược tổn thương mạch máu moyamoya
Ngăn ngừa đột quỵ tái phát và tăng huyết áp
Chỉ định:
- Không triệu chứng
- Đột quỵ thiếu máu nhẹ
- Nguy cơ cao phẫu thuật
Thuốc
Aspirin đến 18 tuổi
Giảm đau: 6.3%
Hạ huyết áp: 5-10%
7.2. Can thiệp mạch
- Tỷ lệ tái hẹp cao
- Có lợi ở những bệnh nhân có phình mạch kèm theo

Hình 5: Điều trị can thiệp trên một bệnh nhân moyamoya cho thấy tỷ lệ tái hẹp cao sau 6 tháng can thiệp mạch.
7.3. Phẫu thuật
Mục tiêu: tăng lưu lượng máu não để giảm nguy cơ đột quỵ
Gồm 2 loại:
- Nối gián tiếp: không tạo miệng nối mạch máu
- Nối trực tiếp
Nối gián tiếp
- Các mô cấp máu như ĐM thái dương nông, Galea, màng cứng, Cơ…
- Tránh được nhiều nhược điểm của tạo miệng nối trực tiếp: nhồi máu do tắc miệng nối, thời gian gây mê dài, đường kính mạch nối quá nhỏ khó thực hiện
- Chỉ định:
- Thường ở trẻ em
- Người lớn moyamoya tiến triển với các động mạch giãn lớn tối đa
Nhược điểm: mất thời gian hình thành các mạch nối không chắc chắn
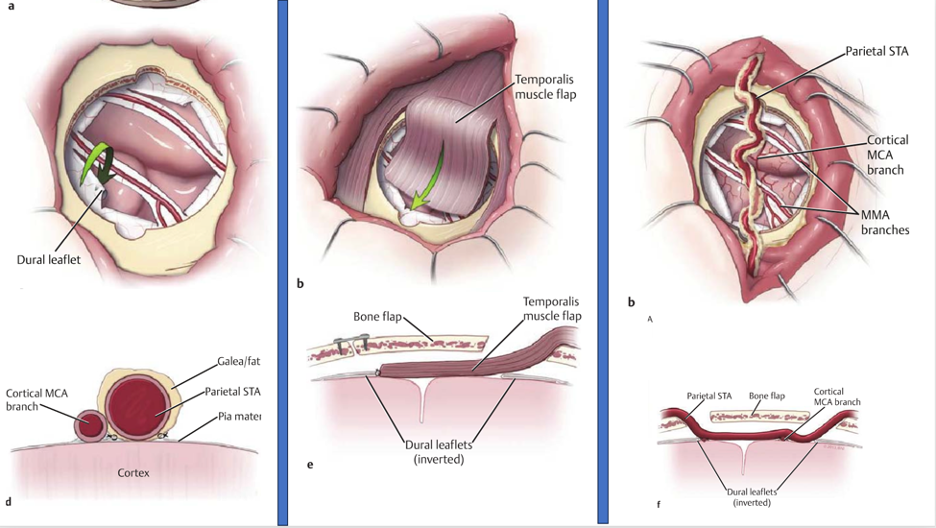
Hình 6: Một số kỹ thuật nối gián tiếp
NỐI TRỰC TIẾP
- Chỉ định
- BN có nhồi máu hoặc xuất huyết có lâm sàng tốt
- Không còn chảy máu
- DSA: Suzuki độ II-IV
- Phẫu thuật > 1 Tháng sau đột quỵ

Hình 7: Kỹ thuật nối trực tiếp tạo nối thông động mạch thái dương nông với động mạch não giữa.
PHỐI HỢP nối gián tiếp và trực tiếp
- TIẾN TRIỂN
Tiến triển tự nhiên của bệnh moyamoya có xu hướng từ từ, ngay cả ở người trưởng thành. Bệnh lý mạch máu thường xấu hơn với sự tắc nghẽn các mạch máu lớn nội sọ và tuần hoàn phụ. Bệnh nhân thường xuất hiện các biểu hiện giảm nhận thức và các triệu chứng thần kinh do đột quỵ thiếu máu não và chảy máu tái phát. Sự đa dạng của hậu quả được xác định bởi các yếu tố sau:
– Trong những nghiên cứu với thời gian theo dõi dài ở những bệnh nhân không được điều trị, tổn thương thần kinh diễn ra từ từ và kết quả tồi được thống kê ở 55-66%.
– Trong một nghiên cứu 40 bệnh nhân người Nhật Bản với bệnh moyamoya không có triệu chứng, phát hiện tình cờ hoặc do sàng lọc, nhồi máu não trên MRI sọ não gặp ở 12 bệnh nhân (30%). Trong nhóm 34 bệnh nhân được điều trị không phẫu thuật và theo dõi với thời gian trung bình là 44 tháng, đột quỵ thiếu máu não và chảy máu trong não xuất hiện ở 1 và 3 bệnh nhân (3%; 9%) tương ứng.
– Moyamoya có thể có tiến triển nhanh hơn và tiên lượng xấu hơn ở bệnh nhân trưởng thành trẻ tuổi so với nhóm bệnh nhân nhi lớn tuổi là kết quả của một nghiên cứu quan sát từ một trung tâm với 204 bệnh nhân Hàn Quốc đã được điều trị phẫu thuật bệnh moyamoya. Nhồi máu gặp nhiều hơn một cách đáng kể ở nhóm bệnh nhân ≤ 6 tuổi khi so với nhóm bệnh nhân > 6 tuổi. Thời gian trung bình từ khi xuất hiện triệu chứng và nhồi máu trước mổ là 3 tháng (1-14 tháng). Tỉ lệ nhóm có kết quả khá trên lâm sàng thấp hơn đáng kể ở nhóm ≤ 3 tuổi khi so với nhóm 3-6 tuổi và ≥ 6 tuổi, chủ yếu là do tình trạng nhồi máu trước mổ.